
- S3-Leitlinie zur Diagnostik, Therapie und Nachsorge für Patienten mit einer chronischen lymphatischen Leukämie(CLL). Online verfügbar unter https://www.awmf.org/uploads/tx_szleitlinien/. 018-032OLl_S3_Chronisch-lymphatische-Leukaemie_2018-04.pdf (letzter Zugriff: 29.09.2020).
- Onkopedia-Leitlinie. Online verfügbar unter https://www.onkopedia.com/de/onkopedia/guidelines/chronische-lymphatische-leukaemie-cll/@@guideline/html/index.html (letzter Zugriff: 29.09.2020).
- International CLL-IPI working group. Lancet Oncol. 2016;17(6):779-790 (und Supplement).
- Campo E et al. Haematologica. 2018;103(12):1956-1968.
- Stilgenbauer S et al. Blood. 2008;112(11):2089.
- Grever MR et al. J Clin Oncol. 2007;25(7):799-804.
- Baliakas P et al. Am J Hematol 2014;89(3):249-255.
- Sharman JP, et al. Lancet. 2020;395:1278-1291.
- Ghia P, et al. J Clin Oncol. 2020;38(25):2849-2861.
- Delgado et al. Am J Hematol. 2017;92(4):375-380.
- https://www.mll.com/erkrankungendiagnostik/chronische-lymphatische-leukaemie-cll/chronische-lymphatische-leukaemie-cll.html (letzter Zugriff: 29.09.2020).
- Döhner H et al. N Engl J Med 2000;343(26):1910-1916.
- Stengel A et al. Leukemia 2017;31(3):705-711.
- Heerema NA et al. Haematol. 2018;212571.
- Jarosova M et al. 2019; https://doi.org/10.1080/10428194.2019.1576038.
- Shanafelt TD et al. N Engl J Med 2019;381(5):432-443.
- Zelenetz AD et al. Lancet Oncol. 2017 Mar; 18(3): 297–311.
- Bond DA, Woyach JA. Curr Hematol Malig Rep. 2019;14:197-205.
- Barf T, et al. J Pharmacol Exp Ther. 2017;363:240-252.
Der Einfluss von Risikofaktoren auf den Therapieerfolg bei CLL
Die klinische Stadieneinteilung nach Rai oder Binet liefert Informationen zu einem möglichen Therapiebeginn sowie zur Prognose der CLL. Mittlerweile haben sich zahlreiche weitere klinische, biologische und genetische Prognosefaktoren etabliert, wobei den genetischen Markern eine zentrale Rolle zukommt.<sup>1,2</sup>
Die klinische Stadieneinteilung nach Rai oder Binet liefert Informationen zu einem möglichen Therapiebeginn sowie zur Prognose der CLL. Mittlerweile haben sich zahlreiche weitere klinische, biologische und genetische Prognosefaktoren etabliert, wobei den genetischen Markern eine zentrale Rolle zukommt.1,2 Nach aktueller Onkopedia-Leitlinie werden IGHV-Mutationsstatus, 17p-Deletionen und/oder TP53-Mutationen sowie komplexer Karyotyp als wichtige genetische Risikofaktoren eingeordnet.1 Vor Therapiestart und während der Therapie werden dementsprechende Testungen empfohlen.2
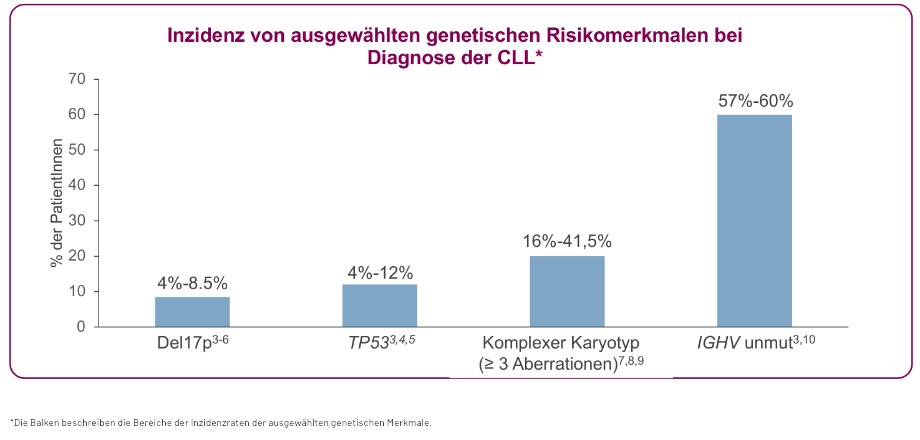
Abb. 1: Die Mehrheit der CLL-Patienten weist bei Diagnose ein genetisches Risikomerkmal auf [3,4,5,6,7,8,9,10]
Die Auswirkung von genetischen Risikofaktoren auf das Gesamtüberleben
Genetische Risikofaktoren können sich nachweislich auf das progressionsfreie Überleben sowie das Gesamtüberleben auswirken (siehe Abbildung 2).7,14,15 Dementsprechend werden in der neuen Onkopedia-Leitlinie nun insgesamt vier relevante genetische Marker als Risikofaktoren angesehen, die für die Therapiewahl mit ausschlaggebend sein können: 17p-Deletion, TP53-Mutation, unmutierter IGHV-Status sowie komplexer Karyotyp. Sie sollten daher vor Einleitung einer Therapie als zusätzliche Diagnostik getestet werden.
Abb. 2: Patienten mit genetischen Risikomerkmalen haben schlechtere Therapieergebnisse [3,7]
TP53-Mutation/17p-Deletion
Für Mutationen im Gen TP53, welches für die Expression des Tumorsupressor-Proteins p53 verantwortlich ist, wurde eine ungünstige Auswirkung auf das Gesamtüberleben und die Zeit bis zur Behandlung festgestellt.11 TP53-Mutationen gehen häufig mit 17p/TP53-Deletionen einher, korrelieren aber unabhängig davon mit einer ungünstigen Prognose.12 Liegt ein TP53-Allel mutiert und das andere deletiert vor, sodass kein funktionsfähiges TP53 gebildet werden kann, führt dies zu einem additiv negativen Effekt.13
Der nachteilige Therapieeffekt von Chemo- und Chemoimmuntherapien bei Patienten mit TP53-Mutation und/oder 17p-Deletion wird in den aktuellen Leitlinien berücksichtigt.1,2 Für diese Patienten-Gruppe werden Therapien mit zielgerichteten Substanzen wie z.B. einem Bruton-Tyrosinkinase-Inhibitor empfohlen.
Aufgrund der klonalen Dynamik der CLL-Zellen ist im Krankheitsverlauf mit dem Erwerb bzw. der Veränderung von Mutationen zu rechnen. Das Ergebnis einer TP53- bzw. einer 17p-Mutationsanalyse vor Therapiestart sollte dementsprechend nicht älter als 12 Wochen sein.1
IGHV-Status
Der Mutationsstatus der variablen Regionen der schweren Immunglobulinketten (IGHV-Status) ist ebenfalls ein wichtiger Prognosemarker, denn Patienten mit unmutiertem IGHV haben nachweislich ein kürzeres Gesamtüberleben (siehe Abbildung 2) und werden früher Therapie-bedürftig.1 Der IGHV-Status ändert sich im Verlauf der Erkrankung nicht und sollte daher einmal vor der ersten Therapieentscheidung bestimmt werden.11 Von den aktuellen Leitlinien wird der IGHV-Status als ein zentrales Kriterium für die Therapiewahl angesehen.14,15
Komplexer Karyotyp
Der komplexe Karyotyp, d.h. drei oder mehr voneinander unabhängige chromosomale Aberrationen, wird mit einer schlechteren Prognose assoziiert und gilt aus diesem Grund in der aktualisierten Fassung der Onkopedia-Leitlinie neuerdings auch als therapieentscheidendes Risikomerkmal.7,14,15
Die Therapiewahl ist entscheidend
In Bezug auf die genetischen Merkmale der Patienten kann die Wahl der CLL-Therapie die Therapieergebnisse entscheidend beeinflussen. Subgruppenanalysen haben gezeigt, dass bei bestimmten genetischen Risikomerkmalen, wie der TP53-Mutation, der 17p-Deletion, dem unmutierten IGHV-Status oder einem komplexen Karyotyp die Prognose für das Gesamtüberleben und das progressionsfreie Überleben unter Chemoimmuntherapien schlechter ist.1,7,16,17 Bruton-Tyrosinkinase-Inhibitoren (BTKi) gehören derzeit zum Standard in der Behandlung der CLL. Unter anderem, da sie auch bei Patienten mit Risikofaktoren gute Therapieergebnisse erzielen - so auch Acalabrutinib, ein BTKi der 2. Generation:18,19 In randomisierten Phase-III-Studien profitierten CLL-Patienten von Acalabrutinib in der Mono- und Kombinationstherapie mit Obinutuzumab, unabhängig von Risikomerkmalen, über alle vordefinierten Subgruppen hinweg.8,9
Quellen
DE-30118